CONSIDER THE LUNG AS A SENSORY ORGAN
At resting, an average person inhales/exhales 5-8 litters of air. This air may vary in oxygen content, carry allergen, pollutants or pathogens. Each of these signals would trigger a specific set of responses. For example, allergen can trigger asthma while intermittent hypoxia can trigger pulmonary hypertension. How the specificity is established is poorly understood.
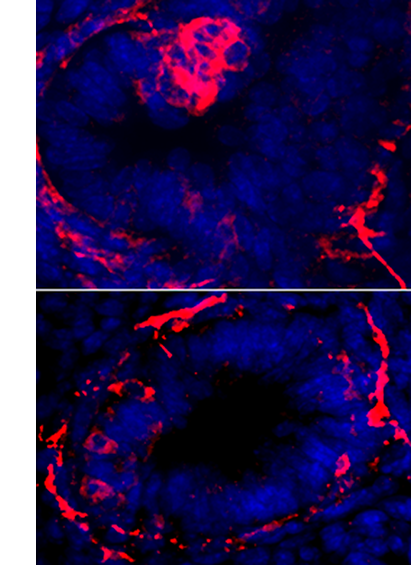
We found that pulmonary neuroendocrine cells, which represents less than 1% of epithelial cells in lung, function as sensors on the airway wall (Branchfield et al., Science, 2016) . These cells can be activated by signals such as allergen, and act through secreting potent neuropeptides and neurotransmitters (Sui et al., Science, 2018). We found that overactivation of these cells leads to heightened immune response at baseline as well as following allergen challenge. Our findings raise the hypothesis that these cells serve as key nodes that mediate lung and immune interactions.
Study of the pulmonary neuroendocrine cells also led us to investigate lung and nervous system interactions, because these cells are innervated. We are mapping the neural circuit that originates from the lung and returns to the lung. Our goal is to understand which aspects of lung function are controlled by the neural circuits, and what underlie the specificity between inputs and outputs.
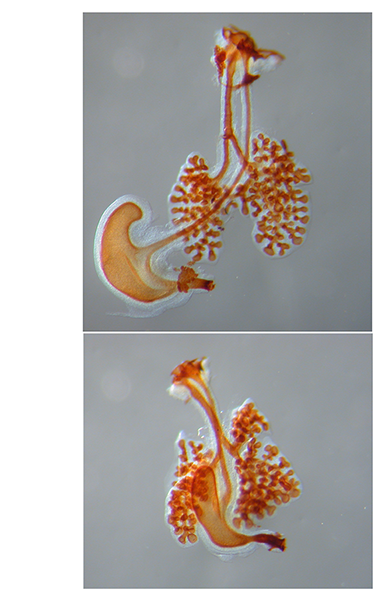
LUNG DEVELOPMENT AND OUTCOMES OF LUNG PREMATURITY
Alveologenesis is the final step of lung maturation, which subdivides the alveolar region of the lung into smaller units called alveoli. Each of the nascent dividers serves as a new gas-exchange surface. Disruption of the final steps of lung development, including alveologenesis results in alveoli simplification, as is seen in premature infants diagnosed with bronchopulmonary dysplasia (BPD). BPD is often associated with lifelong breathing deficiencies, pulmonary hypertension and accelerated decline in respiratory capacity.
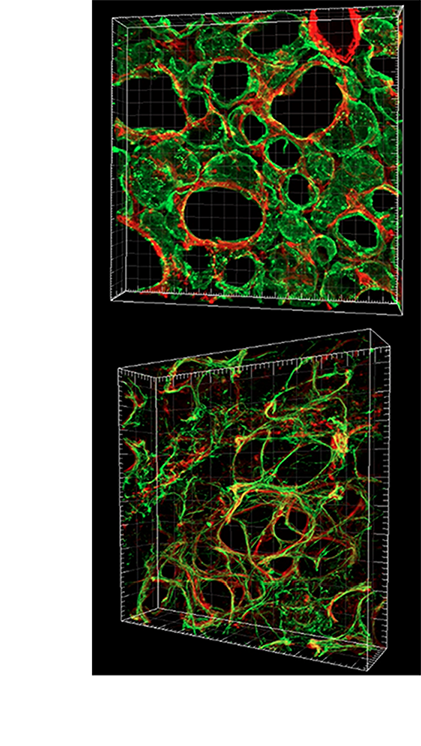
Given that an overarching theme of alveologenesis is thinning and extension of the epithelium and mesenchyme to facilitate gas exchange, we used a three-dimensional (3D) approach to examine the structural architecture and cellular composition of the alveolar cells and extracellular matrix in normal as well as BPD-like mouse lungs (Branchfield et al., Dev Biol, 2015) . We have also used a genetic approach to investigate the genetic mechanism underlying alveologensis (Li et al., Development, 2017 , Li et al., eLife, 2018 , Li et al., JCI, 2020) . Ongoing work in this direction interrogates how different elements of prematurity impacts long-term lung function. The insights revealed by 3D reconstruction of the septae set the foundation for future investigations of the mechanisms driving alveologenesis, as well as causes of alveolar simplification in BPD.
SINGLE CELL AND CRISPR: LUNG DISEASE MECHANISMS
As the acquisition of whole genome sequence data becomes routine, we test the function of candidate patient-specific variants by generating genetic mouse models of disease using CRISPR/Cas9-based genome editing as well as conditional gene inactivation. We have studied a number of lung-related congenital disorders, including tracheo-esophageal fistula (Domyan et al., Development 2011), tracheobronchomalacia (Hines et al., PNAS 2013), and congenital diaphragmatic hernia (CDH) (Domyan et al., Dev Cell, 2013, Branchfield et al., Science, 2016, McCulley et al., JCI, 2018) . Using CDH as an example, we have uncovered different mechanisms from distinct genetic models, suggesting that the nature of the genetic lesions may inform personalized mechanism and treatment. Furthermore, the studies of disease models have also led to exciting basic biology discoveries.
Complementing mechanistic studies using disease models, we are a research center in NHLBI-funded LungMAP consortium to map the single cell transcriptome and epigenome of the lung. Teaming up with The UCSD Center for Epigenomics , we generated single nucleus RNAseq and single nucleus ATACseq data of the human lung from 30-week gestational age, to 3-year old to 30-year old (Wang et al., eLife, 2020). These data are for open access at lungepigenome.org . We are in the process of producing single cell data of diseased lungs, focusing on pediatric lung diseases.